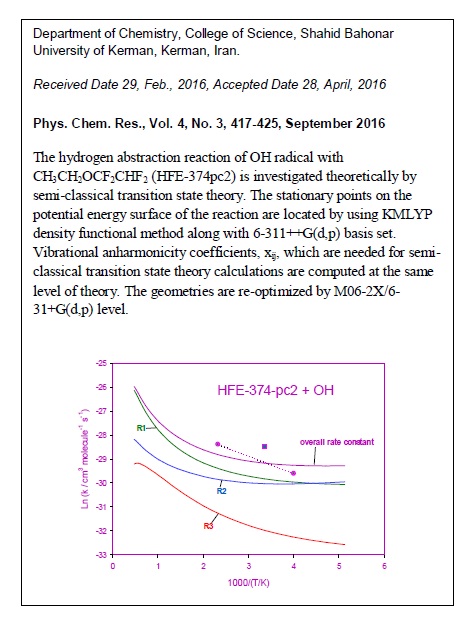
The hydrogen abstraction reaction of OH radical with CH3CH2OCF2CHF2 (HFE-374pc2) is investigated theoretically by semi-classical transition state theory. The stationary points on the potential energy surface of the reaction are located by using KMLYP density functional method along with 6-311++G(d,p) basis set. Vibrational anharmonicity coefficients, xij, required for semi-classical transition state theory calculations, are computed at the same level of theory. The geometries are re-optimized by M06-2X/6-31+G(d,p) level. Single-point energy calculations are carried out by the CBS-Q combination method. Thermal rate coefficients are computed over the temperature range 200-2000 K and they are shown to be in accordance with the available experimental data. On the basis of the computed rate coefficients, the atmospheric lifetime of HFE-374pc2 is estimated to be about 2 months.
Saheb,V. (2016). Ab Initio Theoretical Studies on the Kinetics of the Hydrogen Abstraction Reaction of Hydroxyl Radical with CH3CH2OCF2CHF2 (HFE-374pc2). Physical Chemistry Research, 4(3), 417-425. doi: 10.22036/pcr.2016.14823
MLA
Saheb,V. . "Ab Initio Theoretical Studies on the Kinetics of the Hydrogen Abstraction Reaction of Hydroxyl Radical with CH3CH2OCF2CHF2 (HFE-374pc2)", Physical Chemistry Research, 4, 3, 2016, 417-425. doi: 10.22036/pcr.2016.14823
HARVARD
Saheb V. (2016). 'Ab Initio Theoretical Studies on the Kinetics of the Hydrogen Abstraction Reaction of Hydroxyl Radical with CH3CH2OCF2CHF2 (HFE-374pc2)', Physical Chemistry Research, 4(3), pp. 417-425. doi: 10.22036/pcr.2016.14823
CHICAGO
V. Saheb, "Ab Initio Theoretical Studies on the Kinetics of the Hydrogen Abstraction Reaction of Hydroxyl Radical with CH3CH2OCF2CHF2 (HFE-374pc2)," Physical Chemistry Research, 4 3 (2016): 417-425, doi: 10.22036/pcr.2016.14823
VANCOUVER
Saheb V. Ab Initio Theoretical Studies on the Kinetics of the Hydrogen Abstraction Reaction of Hydroxyl Radical with CH3CH2OCF2CHF2 (HFE-374pc2). PCR, 2016; 4(3): 417-425. doi: 10.22036/pcr.2016.14823